All published articles of this journal are available on ScienceDirect.

Decolourisation of Metal-azo Dyes in Wastewaters by Sodium Peroxodisulphate: A Template for Experimental Investigations
Authors Info & Affiliations
Abstract
Background:
It has long been recognized that the presence of azo metal complex dyes and their non-complexed counterparts in wastewaters, which is due to their manufacturing and use in the textile industries, renders natural waterways intensely coloured, and is therefore aesthetically unacceptable. Azo dye moieties are also known to be precursors of human carcinogenicity.
Objective:
This work aimed at the decolourisation of metal-azo dyes present in wastewaters so that pollution of natural bodies of water can be prevented. Decolourisation is indicative of the destruction of the metal-ligand complex, allowing the retrieval of transition metal ions, which are also water contaminants. Fracturing of the azo bond itself minimizes the potential for the carcinogenicity of these dyes.
Methods:
Decolourisation is achieved by the oxidative action of free radicals furnished by Na2S2O8, the sodium salt of peroxodisulphuric acid (Marshall’s acid). Raman spectroscopy characterizes the dye (ligand) known as “Eriochrome Black T (EBT)” by a peak at 1425 cm-1. Dismantling of its molecular structure by peroxodisulphate will lead to decolourisation accompanied by the collapse of the peak. Concomitantly, as EBT ligands fracture and cease to chelate, metallic ions are released, oxidized to a higher oxidation state, and precipitated as insoluble compounds in alkaline media.
Results:
The concentration of metallic ions in the aqueous phase has been found to be substantially reduced. The successfully treated dye solutions are mostly clear and colourless; their Beer-Lambert absorbances are in the range of 0.02 ≤ absorbance ≤ 0.05. The treatment of Ni(II), Co(II) and Fe(II)-EBT solutions is straightforward; the Cu(II) and Cr(III)-EBT solutions require additional treatment to be included in the above absorbance range. The Cr(III)-EBT is the least responsive to treatment. Fracturing of the azo bond is evinced by Raman Spectroscopy.
Conclusion:
A template to investigate the feasibility of decolourisation of metal-complex dye solutions is pioneered and recommended.
1. INTRODUCTION
1.1. Azo and Azo-metallic Dyes
Dyes and pigments are chemical agents that impart long-lasting colours to clothing and materials. Two distinct structural features that can cause an organic molecule to be coloured are: (1) a colour-bearing group, called a chromophore; (2) an extensive network of alternating single and double bonds (the “conjugation” arrangement) of which the chromophore is a part. Within their chromophoric structures, azo dyes contain one or more azo (-N=N-) linkages with nitrogen atoms, both of which are either bonded to methine groups (-CH=) or directly to aromatic rings (it needs to be noted that the methyne group is not the same as a methylene group –CH2–). The present work concerns the rings present in azo dyes. Azo dyes are intensely coloured because the azo bond between aromatic rings extends the conjugated π-system, resulting in strong absorption characteristics within the visible spectrum. Azo compounds of a wide range of colours have been prepared with various substituent groups on the aromatic rings. They were amongst the first colourants to be used for dyeing cotton and wool.
“Metal-complex dyes” (also called premetallised dyes) are chelates formed by the ions of the transition metals, chromium, cobalt, nickel and copper, with dye molecules as ligands. These colourants are recognized for their lightfastness with predominant application in textiles. There are, however, other uses.
For example, Cu(II) and Pd(II) complexes, with the naturally occurring pigment known as turmeric (a spice from India), as ligands were used in the photodynamic therapy of skin diseases (Franca et al., 2022).
Metal-complex dyes exhibit metal-to-ligand mole ratios mainly of 1:1 and 1:2. Many shades of yellowish-green have been obtained by mixing metal-complex dyes. The transition metals constitute the chromophores in the dye molecules. There are two possible structures for a metal-azo complex (Herbst & Hunger, 2004), as shown in Fig. (1). The nitrogen atom attached to the least nucleophilic aromatic moiety is usually the electron donor to the metal. I and II are fused aromatic rings with attachments, such as hydroxyl, sulphonic, carbonyl, halide, nitro, and amino groups in various positions. Generally, I and II are dissimilar assemblages, but either nitrogen atom can be the electron donor.
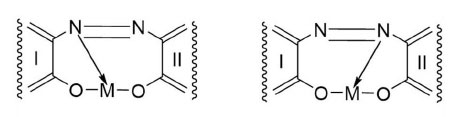
The physicochemical properties of metal-complex dyes are described in encyclopaedic treatises (Hunger, 2003; Zollinger, 2003; Rasik & Oakes, 2014). The electro-analytical chemistry of metal-complex dyes has recently been discussed (Coulibaly, 2021). It can be argued that if the azo double bonds are broken, the aqueous solution of the dye is likely to be decolourised. Therefore, “bleaching” involves the fracturing of the electron-rich azo bonds, and one way to achieve this is the removal of these valence electrons by the S2O82- ion itself and by free radicals made available by thermal homolysis of the S2O82- ion, so that
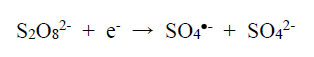 |
(1) |
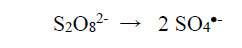 |
(2) |
On reduction, non-toxic sulphate and/or bisulphate anions are formed:
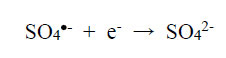 |
(3) |
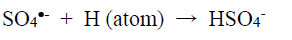 |
(4) |
1.2. Eriochrome Black T (EBT)
Known to the dyeing industry as “Mordant Black 11”, EBT is a mono-azo dye and also a sequesterer of metallic ions. EBT is commercially available as the sodium salt of 3-hydroxy-4-(1-hydroxy-2-naphthylazo)-7-nitro-1-naphthalene sulphonic acid. The EBT anion is shown in Fig. (2) (Çakir et al., 2001). By means of electronic absorption spectroscopy, Hamed et al. established the acid-base equilibrium in an aqueous solution and the azo-hydrazone tautomeric equilibrium in organic solvents. UV-Vis spectral features of solutions of EBT are vulnerable to changes according to the nature of the solvent. For example, Abass and Khadim dissolved EBT in an “aqueous solution of ethanol” to prepare a stock solution for subsequent experiments (involving Et.OH as the minor component). The maximum absorption was exhibited at λ = 567 nm (in the visible region) at this stage. When aliquots of the stock solution were diluted by various amounts of 96% ethanol to make up the solutions for the experiments, the maximum absorption shifted to λ = 508 nm. These solutions were prepared for adsorption of EBT by rice husk waste (in the separation process) in which the EBT moiety does not change; also, assumed constants were the pH and temperature of the surrogate wastewaters. In the present work, the solvent is not inert since a powerful oxidant is dissolved in water, and the pH and temperature are allowed to change with time to mimic a projected Advanced Oxidation Process (AOP) on an industrial scale. With these considerations, the practice of using light absorption maxima at specific wavelengths to track the progress of redox reactions should be questioned and examined more carefully.
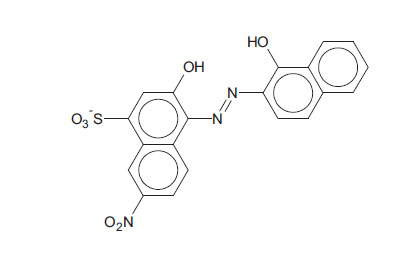
The EBT moiety is triprotic. The sulphonic acid group ionises completely, but the extent of deprotonation of the two hydroxyl groups is a function of pH. With pK2 = 6.3 (dissociation on the right-hand aromatic system) and pK3 = 11.5 (dissociation on the sulfonate ring system) observed at 25oC, EBT has an appreciable solubility of 50 g dm-3 at 25oC (Chemical Book, 2022). To the observer, the colour changes of the indicator system are such that an aqueous solution is red when pH < 6, blue at pH = 7 to 11, and orange when pH > 12.
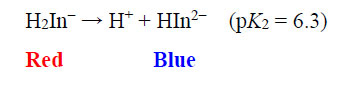 |
(5) |
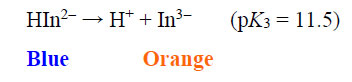 |
(6) |
With a relatively low molecular mass of 461.38 and only one naphthalene group attached to each nitrogen atom, EBT is simple in structure compared to dyes with higher molecular masses, with the mono-azo dye being the most common. Treatment can be implemented for wastewaters containing di- or tri- azo dyes (i.e., dyes with two and three –N=N- linkages per molecule). However, it would not be easy to determine if or which of the azo bridges are fractured. More importantly, EBT is an azo dye that chelates metallic ions, forming compounds of interest in this work. EBT is, therefore, a good model compound for preliminary studies of chemical reactions, leading to decolourisation of azo and metal-azo dyes in wastewaters. EBT has already been used as a model compound in the treatment of surrogate wastewater by using eggshell membranes (Liu, 2011).
Çakir et al. proposed the following structure for the Ni(II)-EBT chelated complex, hereafter designated as the “Ni(II)-EBT complex”, as shown in Fig. (3). They reported the formation constants of two distinct complexes (1:1 and 1:2) to be log10K1 = 8.2 and log10K2 = 11.2, respectively.
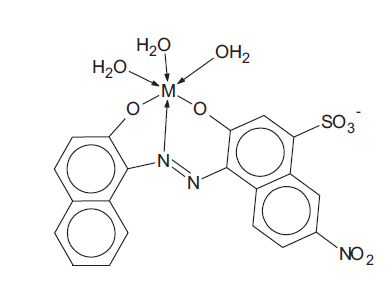
1.3. Toxicology of Aqueous Metal-complex Dye Systems
In a risk assessment, one has to consider the toxicities of both the reactants (metallic ions and un-complexed dye ligands) and the products themselves (the metal-complex dyes) since the dye solution is an equilibrium mixture of all three components. For example, for metallic ion M2+ and ligand L3-, the complex formation is as follows:
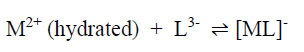 |
(7) |
Formation constants of various metal-EBT complexes have been documented by Masoud. Both unbound metallic ions and free ligands can be present in waste effluents due to unfixed dyes.
There is extensive literature discussing the toxicities of metallic ions and (un-complexed) azo dyes. However, research that addresses the toxicity of metal-azo dye systems is rare, although there is a range of topics within this sparse genre. Cheung and Williams surveyed the literature regarding the toxicities of metal-azo dye systems and noticed that “many researchers are satisfied that metal-azo dyes demonstrate only slight toxicity to humans”, as reported by Brown et al.. Later, Ahn et al. also asserted that they are not the most toxic dyes.
However, the provision of Material Safety Data Sheets (MSDS) in the workplace is mandatory, and Cr(III) dyes are no exception (BASF, 2019). In the USA, the commercial use of metal azo dyes is subjected to the reporting requirements of the Environmental Protection Agency (U.S. Government Printing Office, 2022). In nature, the biological effects of metal-containing dyes on a water flea (Daphnia Magna), which is highly sensitive to water pollutants (Methneni et al., 2021), and on Balb/c Mice (Bae & Freeman, 2007), have been investigated. The substitution and elimination of chromium (and other metals) in dyes have been attempted by many workers (Zarkogianni et al., 2012; Amwele et al., 2013; Kim et al., 2009). The LD50 of un-complexed EBT for rats is 17.6 g/kg (Sigma-Aldrich, 2022).
Three objectives must be achieved in treating wastewaters containing metallic azo dyes:
(1) Decolourise the wastewater.
(2)Retrieve the metallic ions.
(3) Minimise the potential for carcinogenicity by dismantling the dye moiety.
2. LITERATURE SURVEY ON METHODS OF TREATMENT
In their review paper, Cheung and Williams included a section on the treatment of metal-complex dyes based on the overarching theme of extraction of transition metals from their chelated complexes for recycling and reuse; a total of 18 methods were identified. From 2020 to February 2022, a small number of research articles have been published on various techniques for the decolourisation of wastewaters containing non-complexed EBT (Tahir & Yasmin, 2021; Tahir et al., 2022; Brito et al., 2019; Blaisi et al., 2018; Zubair et al., 2022; Moeinpour et al., 2014; Selvi et al., 2020; Selvi et al., 2022; Yusaf et al., 2020). The corpus of knowledge must, however, include the determination of ozonation kinetics of some 1:2 metal complex dyes (Oguz et al., 2008); adsorption of chromium-azo dyes on activated carbon synthesized from tannery wastes (Kebede & Gashaw, 2017); simultaneous adsorption and biodegradation by Aspergillus sp. (Ghosh et al., 2014); adsorption by TiO2 (Wołowicz et al., 2018) and precipitation of dyes by ligand exchange (Pohl, 2020).
Work on decolourisation by peroxosulphates is scarce. Chen et al. decolourised “Acid Orange 7” by oxidation with peroxomonosulphate with the aid of a Co(II) catalyst. In 2008, in her Master’s degree research (supervised by P. Cheung, the principal investigator of the present work), Bruce (2008) successfully decolourised solutions of six dyes (Table 1) by oxidation with the potassium salts of Caro’s acid, H2SO5, and Marshall’s acid, H2S2O8, separately at pH = 2, 7 and 10 and temperatures no higher than 75oC. Later, Zhang et al. decolourised Methylene Blue, Orange G and Direct Blue with peroxomonosulphate at pH = 7.70. Recently, Xiao et al. (2021) decolorized Rhodamine B, Malachite Green, and Methylene Blue by peroxomonosulphate and an iron-based catalyst. No work has been done on the decolourisation of metal-azo dyes in wastewaters by oxidation with sulfur-centered free radicals, such as SO4•-, until now.
|
Dyes (trade Names) |
Chemical Formulae | Molecular Structures | Molecular Masses |
|---|---|---|---|
| Eriochrome Black T (azo dye) CAS 1787-617 |
C20H12N3NaO7S | 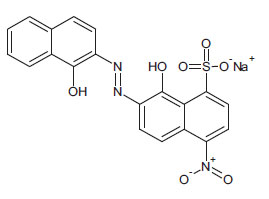 |
461.4 |
| Methyl Orange (azo dye) CAS 547-58-0 |
C14H14N3NaO3S |  |
327.3 |
| Gentian Violet (non-azo dye) CAS 58-62-9 |
C25H30ClN3 | 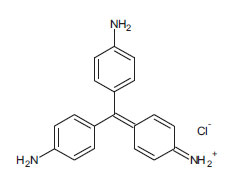 |
407.9 |
| Bromocresol Green (non-azo dye) CAS 76-60-8 |
C19H14Br4O5S | 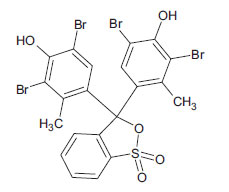 |
698.0 |
| Bromophenol Blue (non-azo dye) CAS 115-39-9 |
C19H10Br4O5S | 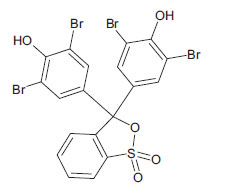 |
669.9 |
| Bromothymol Blue (non-azo dye) CAS 76-59-5 |
C27H28Br2O5S | 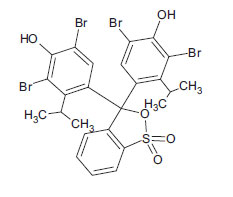 |
624.3 |
3. METHOD OF APPROACH
It must be emphasised that the present work is not an attempt to elucidate the redox mechanisms between SO4•- ions and dyes (both EBT and its metal chelates) at the molecular level. This would require the monitoring of changes in the full spectra (colorimetric data) and absorbance over time to track the course of the chemical reactions with each dye to determine the order of kinetics of every single step. This work is a proof-of-concept to demonstrate the efficacy of peroxodisulphate to decolourize dyes, and whether this can be associated with the cleavage of the azo link in the chromophore.
To add on to the section of methodology, the authors would like to introduce a piece of hitherto unpublished work on attempts to decolourise alkaline solutions of the dyes Gentian Violet (non-azo dye), Bromophenol Blue (BB, non-azo dye), and Eriochrome Black T (EBT, azo dye) by sodium hypochlorite (NaOCl, the chief ingredient of many household types of bleach) in excess at room temperature of 18oC. No transition metal was present in any of these dyes. The results support the authors’ thoughts regarding the general application of Advanced Oxidation Processes (AOP) to the treatment of wastewaters containing dyes. The experiments are detailed in the Appendix of this work. It was intended that observations were to be made by UV-Vis spectrometry and visual inspection. It is worthy of notice that the “absorbance” term in the Beer-Lambert law is the ratio of the intensity of the incident light and the transmitted light; it is a dimensionless quantity and has no units. The three dyes exhibited vast differences; the observations are summarised qualitatively in Table 2.
In the reactions between these dyes and NaOCl, the absorbances observed were only those of single wavelengths (Table 2). In the case of Bromophenol Blue (BB), a rate equation can be written with respect to BB:
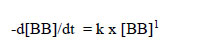 |
(8) |
| Dye | Observations and Results |
|---|---|
| Gentian Violet | All colouration disappeared immediately on mixing the dye solution with NaOCl in a beaker. Clear and colourless water has been the result. The reaction was so rapid that there was no time to introduce a sample of the reaction solution into the spectrometer’s cuvette. |
| Bromophenol Blue | Decolourisation has been attained in 1 hour, and final absorbance has been recorded at 0.08. The peak of the reaction solution at λ = 591 nm did not shift during the course of the reaction. Considering absorbances, the reaction was established as 1st order with respect to the dye (NaOCl in large excess). This classifies the redox reaction as pseudo 1st order. |
| Eriochrome Black T | The orange colouration of the alkaline EBT solution (pH = 12) was found to be absorbed at λ = 605 nm, but the peak shifted to λ = 531 nm on mixing with NaOCl. Another abrupt shift to λ = 395 nm occurred between t = 30 to 35 minutes from the commencement of the reaction. After 1 hour, decolourisation was not complete. A transparent but light yellowish liquid persisted with an absorbance of 0.62. |
The redox reaction between BB and NaOCl proceeds with no complications. Commercially available “HPLC grade” water was the purest water available in the laboratory, and its absorbance was measured by the Perkin-Elmer “LAMBDA 365” UV-Visible Spectrophotometer to be “0.00”, or practically zero. This acts as the baseline for colourlessness. It suffices to treat intensely coloured dye solutions (with absorbances ≥ 1.0) to the extent that they demonstrate absorbances in the range of 0.02 ≤ absorbance ≤ 0.05 (an absorbance of 0.02 means transmission of 1/100.02 = 95% of incident photons). Visual observation may be a subjective criterion of colourlessness but should not be relinquished as a tool for discernment.
As the colouration of the reaction solution becomes less intense, the absorbances of all peaks will decrease towards the baseline absorbance of the purified water in the laboratory used to make up the solution; this baseline is practically “0.0”. Some peaks which appeared earlier in the course of the overall decolourisation reaction may disappear altogether, especially towards the end. The only data which may be needed is a pre-determined value of absorbance (for a production line working end-point) that can indicate the completion of decolourisation, e.g., 0.02 ≤ absorbance ≤ 0.05 (c.f., 0.08 for Bromophenol Blue in Table 2).
However, the question one needs to ask in this preliminary proof-of-concept investigation is whether the collection of large amounts of spectral data would really indicate the verisimilitude of a treatment method. Regardless of whether the shifts in wavelengths were noticed in the course of the overall reaction between Eriochrome Black T and NaOCl (Table 2), it may be necessary to alter the prevailing conditions, namely, an increase in the concentration of NaOCl and the length of time to achieve decolourisation. These parametric requirements can only be discovered experimentally, and at this stage of the investigation, colourimetric data can only be of secondary importance.
With dyes and pigments, there is always the possibility of overlapping spectra to form some type of broad peak. The interference from multiple absorbing species cannot be ignored. A sample of a leaf may, for example, contain a number of chemical species of the green pigment chlorophyll. The different chlorophyll moieties will have overlapping spectra when examined together in the same sample. For a proper quantitative analysis, each chemical species should be separated from the sample and examined individually. The exercise, conducted at room temperature, is challenging enough. To analyse samples taken at different temperatures with the intention of correlating them will be difficult. One has to ask why this needs to be done.
What is important is that one should always establish empirically (possibly in a pilot plant facility) as to how long it takes to achieve decolourisation, given a set of physicochemical conditions, and whether this time scale is acceptable if carried out on an industrial scale. Peroxodisulphate ions are a powerful oxidizing agent regardless of media pH (Bruce, 2008). This is an advantage to the wastewater treatment process because no pH adjustment at any stage is necessary. The two parameters available for control at the outset are: (1) the concentration of the oxidant (up to its solubility limit) and, therefore, the oxidant-to-reductant mole ratio; (2) and the temperature at which the redox scheme proceeds. Usually, increasing the temperature and the concentration of the oxidant will shorten the time required for the reaction. As an example, Bruce (2008) discovered that the three non-metallic dyes in Table 2 will not be decolourised by HSO5- nor S2O82- (2 wt.% solution) at room temperature, but the decolourisation of 0.1 mM solutions proceeds to completion within 35 minutes when the reaction solution is heated to the temperature range of 65 - 75oC, regardless of the pH of the reaction solution at the commencement of the redox reactions. In fact, the pH values of all solutions containing peroxodisulphate fall with time due to the following two reactions, as reported by Ahmadi et al. Therefore,
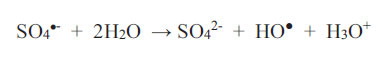 |
(9) |
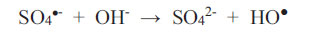 |
(10) |
As sulphate radicals SO4•- are formed according to Equations 1 and 2, they react with water to produce hydroxonium ions (Equation 9), and they also consume hydroxyl ions (Equation 10). Both reactions furnish HO● radicals. Since EBT is an indicator dye, the colouration will shift from orange to blue and then to red as the chemical environment changes from alkaline to acidic. This continuous shift in the wavelength of maximum absorption in the visible range is not difficult to monitor if it were not for the attack on the dye itself by the oxidant (which may transform it by intramolecular electronic rearrangement and/or fracturing it). It is not known what the intermediate products are and what the changes in colour represent. The mechanistic pathway to decolourisation is not expected to be simple and neither is the interpretation of the spectral features representing dynamic and complex molecular events.
Reactions between S2O82- and H2O have been reported at 50oC (Kolthoff & Miller, 1951), but rapid decomposition of S2O82- to furnish SO4•- at higher temperatures in the range 60-90oC has also been established (Bruce, 2008; Quig, 2015). An indication that sulphate radicals have been produced is the accompanying rapid drop in pH (Equations 9 and 10). Usually, a reaction mixture is heated up from room temperature to a set temperature in order for reactions between SO4•- and target species to take place.
It has never been advisable to introduce a hot liquid into a cuvette (regardless of glass or quartz) as this may irreversibly alter the dimensions of the cuvette, rendering all subsequent measurements imprecise. In order to introduce a sample of a heated reaction solution into a cuvette, it is essential to cool the solution to room temperature with tap water, but this is unsatisfactory. The explanation is as follows. Suppose one wishes to examine the overall UV-Vis spectrum of a reaction solution at 50oC. Since it is undesirable to introduce hot liquid into a cuvette, many experimenters will start to cool the glassware containing the reaction solution under a tap to room temperature, about 20oC. While the solution is cooling, the reaction slows down since the rate of reaction is a function of temperature, but the reaction does not stop just because the temperature of the solution is dropping. Reaction still proceeds, only slower. So, when the temperature reaches 20oC, the UV-Vis spectrum will not be the same as that of the solution at 50oC, but it will represent the summation of reaction events occurring between 50oC and 20oC. Any significant changes in spectral features between 50oC and 20oC, which no doubt there will be, are missed. The higher the temperature attained (beyond 50oC), the longer it takes for the solution to cool to room temperature, and the further the entire scheme of redox reactions may develop. Thus, to establish continuity between individual spectral graphs will be difficult.
A word of caution needs to be said about unexpected effects, which may lead to erroneous absorbance readings. It is possible that in the initial stages of treatment of an alkaline solution of a metal-azo dye before the acidifying effects (portrayed in Equations 9 and 10) suppress pH values to below 7, there may be competition between OH- ions and dye ligands for the transition metallic ions Mn+. Therefore, insoluble metallic hydroxides M(OH)n may coagulate and disperse to become a colloidal suspension depending on the solubility product of the metal hydroxide and the settling time of such solid particles. The Tyndall effect, which is photon scattering by suspended particles in liquid samples, may cause serious errors in the measurement of Beer-Lambert absorbance. The presence of particles in the cuvette will scatter light and render results irreproducible.
The use of Raman spectroscopy in this work did not mean to replace UV-visible spectrophotometry as an instrumental method of analysis, as the two methods aim to obtain different types of data. The azo group –N=N- in various molecules is easily identified by specific wavenumbers (in the unit of cm-1). The aim is to witness the peaks of these characteristic wavenumbers collapse as an indication of the breaking of the azo bond and, therefore, of the chromophore at the end of the experiment. EBT has not been classified as a carcinogen, but the ability to break the azo bond by electron-transfer reactions during wastewater treatment has implications for the detoxification of carcinogenic aromatic azo dyes.
4. EXPERIMENTATION AND RESULTS
Two sets of experiments (A and B) were performed. Set A explored the decolourisation phenomena of EBT alone since the free ligands also constitute the dye mixture (Equation 6). Operating conditions established in Set A were then applied to the decolourisation of various aqueous solutions containing metal-EBT complexes, which comprised the Set B experiments.
The following chemicals/materials were obtained from Sigma-Aldrich (Gillingham, Dorset, UK), and were of pure analytical grade: Eriochrome Black T, chromium (III) nitrate nonahydrate, iron (ii) chloride tetrahydrate, cobalt (ii) sulfate heptahydrate, nickel sulfate hexahydrate, copper (ii) sulfate pentahydrate, sodium thiosulphate pentahydrate, sodium persulfate (Na2S2O8, 98% purity), and HPLC grade water (CAS: 7732-18-5). An ion-exchange resin branded as Amberlyst-15 (CAS: 39389-20-3) was also obtained. “Activated granular carbon” (10 – 18 mesh, 2.06 mm down to 1 mm sieve size) was obtained from VWR (Lutterworth, Leicestershire, UK). The filter papers used were Sigma-Aldrich’s reinforced membrane, mixed cellulose esters, 0.2 µm, and polyester web 293 mm.
4.1. Set A – Determination of Operational Parameters of Decolourisation of Un-complexed EBT
4.1.1. Decolourisation of a Solution of Free EBT Ligands and Minimization of Oxidant Usage
An attempt was made to decolourize a solution of EBT within two hours at a temperature not higher than 70oC. A stock solution of EBT was prepared by dissolving 0.420(3)g of EBT powder in 2 dm3 of deionised water, resulting in a molar concentration of 5 x 10-4 mol dm-3 (0.5 mM).
Another EBT solution was prepared by adding 50 cm3 of deionised water to 100 cm3 of the stock solution. The concentration of EBT was then given by (100/[50 + 100]) x 0.5 = 0.33 mM. This was labelled as Solution “A”. The pH of solution “A” was measured by a calibrated pH meter and was found to be 5.4(0). To 100 cm3 of Solution “A”, 0.3 g NaOH was added, raising the pH to 11.8, resulting in a translucent, deep dark orange solution. This step to render the medium alkaline was necessary since the dye coagulates in acidic environments and the resulting coagulates settle. 4.0 g of Na2S2O8 was then dissolved in solution “A” (4% w/v with respect to the oxidant), and the pH before the initiation of redox reactions by heating was measured to be 10.9. Aqueous solutions of Na2S2O8 (as the only solute) are acidic, and without the pH adjustment mentioned above, any addition of Na2S2O8 to an EBT solution that is slightly acidic will render it even more so; EBT will precipitate, and the experiment will not proceed.
The reaction solution was heated by a magnetic heater-stirrer in a fume cupboard. The room temperature was 20°C. As the solution was heated, the temperature increased by 2°C per minute. A distinct change of colouration from deep dark orange to dark brown occurred at 40°C, then to bright orange at 45°C, and then to an even lighter shade of orange at 60°C. As soon as the temperature reached 65°C (i.e., after about 22 minutes of heating), the solution appeared to be clear and colourless. The decolourised solution was kept at 65°C for 1 hour in a water bath to eliminate vestigial remnants of the dye populace, in order that data could be collected for comparison with those representing the decolourisation of metal-EBT solutions. The choice of “4 g” (4% w/v) of Na2S2O8 was predetermined by the following optimisation exercise. A range of masses of Na2S2O8 from 1 to 10 g were dissolved in solutions “A” and subjected to the heating procedure outlined above. This was an exercise to discover whether the oxidant applied was inadequate or in excess.
4.1.1.1. Results and Discussion
Clear and colourless liquor resulted after treatment of the EBT solution with Na2S2O8 (4 wt. %) as an oxidant and heating the reaction solution to 65°C. There was no sludge formation observed. Regarding the task of minimization of oxidant usage, observations of the appearance of the treated waters are tabulated in Table 3.
Table 3.
| Test solution no. | Na2S2O8 applied (g) | pH (before heating) |
Observation of treated water |
Comments |
|---|---|---|---|---|
| 1 | 10.01(3) | 9.4 | Clear and colourless solution. No sludge formation. |
Oxidant applied was in large excess. |
| 2* | 5.00(2) | 10.2 | Clear and colourless solution. No sludge formation. |
Oxidant applied was in excess of about 25%. |
| 3 | 4.01(6) | 10.9 | Clear and colourless solution. No sludge formation. |
“Optimal” concentration was established with this data point. |
| 4 | 3.04(4) | 11.6 | Transparent, very pale orange. No sludge formation. |
Inadequate oxidant. |
| 5 | 2.01(0) | 12.1 | Transparent and bright orange. No sludge formation. |
Inadequate oxidant. |
| 6 | 1.03(7) | 12.3 | Transparent and bright orange. No sludge formation. |
Inadequate oxidant. |
The minimum amount of Na2S2O8 needed to decolourise an EBT solution by heating to 65°C was determined to be in the range of 3.044 to 4.016 g (per 100 cm3 solution), i.e., 3% w/v < (% w/v)min ≤ 4% w/v. No precipitate of sludge was formed in any of the above-treated waters.
4.1.2. Control Experiment to Determine Whether Heating Alone will Decolourize EBT
Step 1: 10 cm3 of Solution “A” (0.33 mM of EBT) was diluted 25 times with deionised water in a 250 cm3 volumetric flask, with no peroxodisulphate present; this solution is labelled as Solution “B”. The pH was then raised to a value of 12. The total absorbance of Solution “B” was noted to be 0.334. Solution “B” was not heated but allowed to stand on the bench for observation.
It was necessary to dilute solution “A” because its absorbance (measured to be 4.0) was far too high. This means that the fraction of photons detected would only be 1/(104) ≈ 0.01% of all incident photons, which is too low to make meaningful comparisons if the action of heat alone causes only minute changes in the overall intensity of the colouration of the solution. For precision, absorbencies should be no higher than 1.0, corresponding to a 10% transmittance of incident photons (Owen, 2000).
Step 2: 100 cm3 of Solution “B” was then subjected to the heating program outlined in section 3.1.1 above, but in the absence of peroxodisulphate. After cooling to room temperature, the absorbance of this solution was measured and compared to that of the unheated solution “B”. One must not transfer a hot liquid to the glass or quartz cuvette (of 1 cm path length), which is the cell holding the sample, as this may change the dimensions of the cell irreversibly.
4.1.2.1. Results and Discussion
Absorbencies of the unheated and heated solutions were observed to be 0.334 (at the 550 nm broad peak) and 0.1910 (at the slightly shifted 546 nm broad peak), respectively, with sharp and discernible peaks in the UV-Visible spectral profile of the heated solution. Heating resulted in a reddish-orange solution. The EBT moiety may have undergone structural transformation by the action of heat. The change in molecular structure may involve fragmentation or not. The heated solution remained visibly coloured after cooling to room temperature when the absorbance reading was taken. Clearly, decolourisation to obtain clear, transparent, and colourless water was not due to the action of heat alone but to oxidation by Na2S2O8.
4.1.2.2. First Attempts at Operational Parametric Optimisation
In a separate experiment of theoretical interest, an attempt was made to discover how long it will take to decolourise solution “B” with no heating (i.e., isothermally at 18oC) and with half the “optimal” concentration of the applied oxidant Na2S2O8, i.e., 2 wt.% (0.084 M) instead of 4 wt.% which is the concentration used in Test Solution no. 3 in Table 3. It was found that Solution “B” decolourised after 2½ days (continuous stirring). Therefore, Na2S2O8 (0.084 M) managed to decolourise EBT (0.33 mM, i.e., 3.3 x 10-4 M) at 18°C after 2½ days. The mole ratio of oxidant-to-reductant is 0.084/0.00033 = 255:1, but it should be noted that there is almost no change in the wastewater volume when Na2S2O8 (s) is dissolved in water even as the oxidant exists overwhelmingly. Hypothetically, if there is ample liquid storage (tank) space in an industrial installation and if copious time is allocated for processing wastewaters, then all that is required is a minimum amount of oxidant. Not many factories have those facilities.
A less-than-ideal situation is when NaOCl is chosen as the oxidant. When an aqueous solution of an oxidant is added to wastewater, the overall volume of liquid to be managed increases; moreover, hypochlorite cannot be added to acidic wastewaters (without pH adjustment to the alkaline regime) as this will cause green chlorine gas to be evolved, thereby negating its use on health and safety grounds.
4.1.3. Raman Spectroscopy of EBT Solution
Raman spectra of Solution “A” (0.33 mM) and of the peroxodisulphate-treated, clear, colourless and transparent solution were obtained and shown in Fig. (4).
Laser Raman micro-spectroscopic analyses were carried out using a Renishaw RM-1000 Raman Spectrometer, which is equipped with a thermo-electrically cooled charge-coupled device for light detection and a 513.5 nm argon ion laser. The instrument was calibrated daily with a silicon standard at 521 cm-1. The system was operated in confocal mode with the laser focused on the sample through the x50 (long working distance) objective lens of the Olympus Model BH-2 petrological microscope. Samples were held in simple open glass capillary tubes, and to avoid heating of the sample by laser photons, the power to be received by the sample was set at a value of 1 x 10-3 Watt maximum. Spectra were recorded over the range 250 cm-1 to 3750 cm-1 using 3-minute scans, resulting in a total exposure time of about 20 minutes per sample. All analyses were conducted at room temperature since the subject of interest was the cooled final effluent and whether or not it could be discharged to sewer. Peak-fitting was performed using Galactic GAMS/32 software, which uses mixed Gaussian-Lorentzian curves.
4.1.3.1. Results and Discussion
The azo –N=N- symmetrical valence stretch observed at 1425 cm-1 is closest to 1440 cm-1 in literature (for a trans-isomer, while the stretch for the cis-isomer is located at higher frequencies), while examples of individual peak values for various azo dyes fall in the range 1408 to 1435 cm-1 (Trotter, 1977). A broad band at 1380 – 1450 cm-1 was also reported for the same stretching mode (Vandenabeele et al., 2000); therefore, a peak observed at 1400 cm-1 in the obtained spectrum fell within this range.
In the lower spectrum shown in Fig. (4) (beneath the “colourless wastewater” label), not a single Raman peak can be seen, suggesting that the N=N bond was broken. In fact, the entire spectrum that had been previously observed for the untreated dye solution simply collapsed.
Sulphate free radicals SO4•- obtained by the homolytic fission of S2O82- have the ability to extract electrons from the N=N bond. The fracture results in the discontinuation of the π-conjugation system responsible for colouration. Since the EBT moiety is not symmetric, one would expect the N=N stretch mode to be active both in infrared (IR) and Raman.
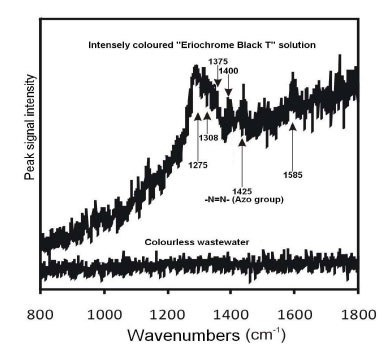
Due to the high absorption of the EBT dye solution, even at low concentrations, it can be hypothesized that a resonant Raman Effect enhanced the spectral intensity. This is a quantum-mechanical effect whereby the Raman scattering cross-section of a molecule is increased when an absorption band is close to the absolute frequency of the exciting photon. If Raman resonance was indeed the reason for the measurable intensity of the dye solution, then it is easy to understand why no peaks were observed in the decolourised solution. While it is reasonable to assume that the concentration of organic fragments bearing the C=C, C-H, and C=O groups in the decolourised solution was similar to that in the original dye solution, one would expect to see some peaks in the decolourised solution in the absence of any resonant enhancement. However, the removal of conjugation by “bleaching” destroyed any resonant enhancement, and so reduced the overall Raman intensity of the decolourised solution, perhaps to the point of being undetectable. It may be possible to identify the residual organic molecules in the decolourised solution by LCMS (Liquid Chromatography-Mass Spectrometry) subsequent to the removal of Na2SO4 by crystallisation. The SO42- ions are produced from single-electron transfers to the free radical, as shown in Equation 3.
The azo dye EBT has never been classified as a carcinogen, but the general understanding of the formation of carcinogenic aromatic amines is such that they can be formed when azo dyes are reduced by coming into contact with human tissues (Pinheiroa et al., 2005; Nguyen & Saleh, 2016). More azo dyes are continually being listed as carcinogenic (Government of Australia, 2020).
In industrial wastewater treatment, sulphate radicals can extract electrons from the azo bond, thereby breaking it, with the result that the possibility of hydrogen addition is much reduced. Any added hydrogen atom may also be abstracted by the radicals in the highly oxidising environment. An interesting case arises when –NH2 groups are present on benzene rings in a di-azo dye, such as “Direct Blue 14”, whereby the breaking of both azo bonds leads to the formation of the aromatic amine o-tolidine (Platzek et al., 1999). Whether an advanced oxidation process (AOP) can oxidize amine groups to nitro groups with the fragmentation of the aromatic amine requires further experimentation and chemical analysis.
4.2. Set B - Decolourisation of Metal-EBT Dye Solutions
The description of Set B experiments is organised in the following way:
4.2.5. Decolourisation of the Cr(III)-EBT dye solution.
Each of the above subsections is divided further. For example, with the Ni(II) dye, after depicting the experimental details, the interpretation of results then follows.
Results and Discussion:
(a) Observations during decolourisation.
(b) Metal (nickel) recovery.
(c) Control experiment and inferences.
The same layout is repeated for the Co(II), Fe(II), Cu(II), and Cr(III) dyes.
4.2.1. Decolourisation of the Ni(II)-EBT Dye Solution
Mole ratios of metal-to-ligand, namely, 1:1 and 1:2, are known for metal-azo complexes (Liu, 2011). In this exercise, excess EBT is used in some industries, whereby excess ligands are applied to “ensure complexation”. Excess ligands, unfortunately, are a sacrificial cost unless they can be recycled for reuse; otherwise, they will consume more oxidant than necessary during wastewater treatment. In this exercise, dye solutions with a mole ratio of 1:4 are prepared for each of the metallic ions Ni2+, Co2+, Fe2+, Cu2+ and Cr3+. The solutions are prepared simply by mixing the required stoichiometry of the ingredients. All solutions appear deep dark blue and opaque before treatment. Na2S2O8 is added to each of these solutions to oxidize the metal-EBT complexes.
To 100 cm3 of the EBT stock solution, 50 cm3 of the solution containing the free (i.e., non-chelated) metal ion was added. The concentration of EBT in the reaction solution was then given by (100/[50 + 100]) x 0.5 = 0.33 mM. Stock solutions of sulphate salts of the free metallic ions Mn+ were prepared at a concentration of 0.25 mM. All of these solutions before the commencement of chemical reactions in peroxodisulphate exhibited absorbances greater than unity, all in the close vicinity of 1.10.
Subsequent to mixing with the EBT solution, the concentration of Ni2+ in the reaction solution was (50/150) x (2.5 x 10-4) = 8.33 x 10-5 mol dm-3. The mole ratio of ligand-to-metal was [EBT3-]:[Mn+] = 3.33 x 10-4 / 8.33 x 10-5 = 3.997 ≈ 4. The same ratio applies separately to solutions containing Co2+, Fe2+, Cu2+ and Cr3+ ions. The volume of the prepared Ni(II)-EBT solution was 150 cm3 with pH = 4.3, and this was divided into two portions: (a) To 100 cm3 of it, 0.3 g of NaOH was added and pH increased to 12.1. On dissolving 4 g of Na2S2O8 in this alkaline solution, pH dropped to 10.9 (but the aqueous mixture remained in the intended alkaline region so that EBT would not coagulate and settle). We called this Solution B-1. (b) The remaining 50 cm3 was Solution B-2 (without Na2S2O8) that underwent a control experiment.
Solution B-1 was heated from room temperature of 20oC to 65°C (this took approximately 22 minutes), and this temperature was maintained for 1 hour in a water bath. Then, 5 cm3 of the treated (then cooled) solution was diluted tenfold in a 50 cm3 volumetric flask.
To establish the effect of heat alone without the presence of any oxidant, a control experiment was carried out with Solution B-2 of volume 50 cm3 in 2 steps:
Step 1: 5 cm3 of Solution B-2 was diluted 25 times in a volumetric flask, and the absorbance was obtained. There was no heating conducted on this solution.
Step 2: 45 cm3 of Solution B-2 was heated. Then, 5 cm3 of this heated solution was diluted 25 times, and the absorbance was also obtained.
4.2.1.1. Results and Discussion
(a) Observations during decolourisation. When the temperature of the reaction solution reached 40°C, the water became clear, transparent and orange (akin to solutions of Na2Cr2O7). When the temperature reached 57°C, the solution turned dark brown, but remained clear (no precipitate at this point). At 65°C, a clear, colourless liquid resulted, and a black precipitate was formed on settling. Profuse effervescence of gas bubbles was evident after the appearance of the black precipitate, which is likely to be the Ni(III) compound Ni.O(OH) (definitely not the green gel Ni(OH)2), and the Ni(III) compounds can potentially split water (Ren et al., 2015).
Subsequent to the dismantling of the dye ligand to the extent that it loses its ability to sequester metal ions, Ni2+ ions released from the chelated complex into aqueous solution are oxidized to the +3 state and precipitated as black amorphous solid particles in an alkaline and strongly oxidizing environment. The precipitation is stoichiometric, and the overall redox reaction is as follows:
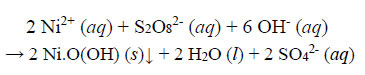 |
(11) |
As the reaction mixture was kept at a temperature of 65°C in a water bath for one hour subsequent to these events, no visible changes were observed during this hour. Instead of waiting for the particles to settle, a filter paper was used to separate the black particles from the aqueous suspension. The filter paper was Sigma-Aldrich’s “reinforced Membrane, mixed cellulose esters, 0.2 µm, and polyester web 293 mm”. The final absorbance of filtrate at room temperature of 18oC was 0.025.
(b) Nickel recovery. The treated solution was filtered to give clear, colourless, and transparent water. The analysis of this colourless water was carried out by using ICP-OES (inductively coupled plasma-optical emission spectrometry); the analytical system used was the HORIBA Jobin Yvonne Ultima 2C model, which showed the concentration of nickel ions to be 0.053 ppm (0.9 μM). The percentage recovery of nickel was calculated to be [1 – (0.9/83.3)] x 100% = 99.0%.
Vestiges of Ni(II) ions that still exist in the treated solution can be removed by ion exchange. Before the discharge of satisfactorily treated waters into public utilities, the pH has to be adjusted close to neutrality. At pH = 7, the speciation of Ni(II) ions includes not only the [Ni(H2O)6]2+ ion but also the binuclear [Ni2(OH)]3+ and the polynuclear [Ni4(OH)4]4+. These species were reported by Burgess (1978). Regardless of laboratory readings, industries involving electroplating operations routinely install cation exchange columns to lower the concentration of metal ions below discharge limits. Had Ni(II) ions been trapped in anionic complexes subsequent to wastewater treatment, they can be removed by anion exchange columns. It also needs to be noted that sulphate ions are benign and ubiquitous in nature, and can be discharged into public utilities.
(c) Control experiment and inferences. The control experiment was performed in exactly the same way as that for the pure EBT case, at the end of which, the water phase was observed to be transparent but reddish brown. Therefore, heat alone did not decolourize the dye solution and helped to lighten the intensity of colouration at best. Aggregates of brown suspended solids (2.5 mm) settled on standing, but these were not equivalent to the black solids suspended in the liquid solution that had been fully treated by Na2S2O8.
4.2.2. Decolourisation of the Co(II)-EBT dye solution
The Co(II)-EBT solution was subjected to the same processing conditions and procedure as that of Ni(II)-EBT above.
4.2.2.1. Results and Discussion
(a) Observations during decolourisation. The observations and end results of the decolourisation of the Co(II)-EBT complex were also very similar to those of Ni(II)-EBT, with the same sequence of events evinced. The Co(II)-EBT dye solution was decolourised by Na2S2O8. Gas evolution was also apparent after the appearance of a black suspended solid of a Co(III) compound, likely to be Co.O(OH), which settled. The final absorbance of the treated solution after filtration at a room temperature of 18oC was 0.024.
The chemistry was very similar to that of the oxidation of the Ni(II)-EBT dye. Subsequent to the decomposition of the dye ligand, Co2+ ions released from the chelated complex oxidized to the +3 state and precipitated as black amorphous Co.O(OH) particles. The precipitation was stoichiometric; the overall redox reaction is as follows:
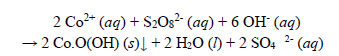 |
(12) |
(b) Cobalt recovery. The final concentration of cobalt ions in the treated solution was 0.048 ppm (0.81 μM). The recovery of cobalt ions from the solution was [1 – (0.81/83.3)] x 100% = 99.0%.
As in the case of nickel as above, any Co(II) ions that still exist in the solution can be removed by ion-exchange columns if desired. As in the case of wastewaters containing nickel ions described above, the pH of treated liquors is adjusted close to pH = 7 before discharge. In very weakly acidic solutions, species, such as the dimer [Co(OH2)Co]4+, exist (Burgess, 1978), besides [Co(H2O)6]2+. The argument about whether similar dimers exist for Co(III) ions continues.
(c) Control experiment and inferences
In the control experiment, it was discovered that heat alone did not decolourize the solution but lightened the colouration. A brown sludge precipitate also made its appearance.
4.2.3. Decolourisation of the Fe(II)-EBT Dye Solution
The Fe(II)-EBT solution was subjected to the same processing conditions as that of the nickel and cobalt EBT dyes above.
4.2.3.1. Results and Discussion
(a) Observations during decolourisation. Crystals of FeCl2.4H2O do not dissolve in water immediately. To solubilise them, water was first added to a mortar, followed by the crystals. Grinding produced a fine, murky suspension (if water had not been added for the grinding, the resulting powder would have stuck to the mortar, and it would have been very difficult to remove). This suspension was washed into a 1 dm3 volumetric flask with 500 cm3 of water, with the aid of a glass filter funnel. 2 cm3 of HCl (1 mol dm-3) was added to keep the solution acidic, and the solution was swirled until the suspended particles dissolved. The flask was then topped up with deionised water to the 1 dm3 mark. The final pH of the prepared solution was measured to be 2.70. At this pH, Fe2+ formed a slight pink complex with EBT, unlike the bold reddish nickel and cobalt complexes in an acidic solution. The addition of 0.3 g NaOH turned the solution orange, with a resulting pH of 12.0.
Decolourisation of the complex in the reaction solution was the most rapid of all the metals studied. Decolourisation was practically completed 15 minutes after heating commenced, when the temperature of the reaction solution rose from 20oC to 50°C. The final absorbance of the treated solution after filtration at a room temperature of 18oC was 0.022. The redox reaction between ferrous ions and persulphate subsequent to the decomposition of the EBT ligand is presented as follows:
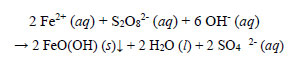 |
(13) |
(b) Iron recovery. The reddish precipitate obtained was a monohydrate with empirical formula FeO(OH).H2O, and it has always been conveniently expressed as “iron (III) hydroxide, Fe(OH)3”. The residual iron content in the treated solution was 0.039 ppm (0.69 μM). Therefore, iron was recovered at [1 – (0.69/83.3)] x 100% = 99.1%.
There were hardly any Fe(II) ions remaining in the solution under the processing conditions, and Fe(III) species predominated. The aqua ion [Fe(H2O)6]3+ was present. The experiments in this work were carried out in the fume cupboard, and air alone oxidized Fe(II) to Fe(III) with ease. There is a lot of instrumental analytical evidence that suggests the existence of the species [Fe2O]4+ and [Fe2(OH)2]4+ in solution, and is documented in the work done by Burgess. Iron rust is a gigantic network of three-dimensional polymers. A cation exchange matrix will remove these ions from the solution.
(c) Control experiment and inferences. The control experiment showed that heating alone could not decolourise the metal dye solution in the first 15 minutes of heating, merely lightening the colour intensity.
4.2.4. Decolourisation of the Cu(II)-EBT Dye Solution
The Cu(II)-EBT solution was subjected to the same processing conditions as those of the metal-EBT dye solutions discussed above.
4.2.4.1. Results and Discussion
(a) Observations during decolourisation. In the first 22 minutes of heating starting from a room temperature of 20oC, the reaction solution changed from deep orange to light orange at 60°C, and then became peach-coloured at 65°C. During the next 45 minutes of immersion in the water bath, this peach colour faded rapidly, leaving the solution with a very light tint of peach colour at the end of the entire treatment. A black precipitate of CuO was formed. Cu(II) could not be oxidized to a higher oxidation state, and the black precipitate of CuO formed was simply a dehydration reaction upon warming, therefore:
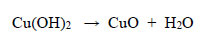 |
(14) |
After cooling the treated solution to room temperature and filtering, the liquid was subjected to a sorption process to eliminate the last vestiges of peach colouration.
Step 1: 2 g of activated charcoal was added to the solution and stirred for an hour in the beaker. The charcoal used was “activated granulated carbon”, 10 - 18 mesh. This removed the peach tint. A colourless liquid thus has been the result.
Step 2: The colourless solution was acidified to pH = 2.0 to convert any Cu(OH)42- ions to Cu2+ ions. Then, Cu2+ ions were removed via ion exchange by adding 0.2 g ion-exchange beads (Amberlyst-15) and stirring the liquid for 30 minutes. The final absorbance of this liquid after filtration was obtained as 0.036.
(c) Copper recovery. ICP analysis of the final liquor determined the residual [Cu2+] = 0.55 ppm (8.7 μM). Therefore, the recovery of copper achieved was [1 – (8.7/83.3)] x 100% = 89.6%.
As in the cases of cobalt, nickel and iron, the pH of industrial wastewaters containing copper ions is adjusted close to neutral before discharge into public utilities. At neutral and slightly acidic pH values, the ion [Cu(H2O)6]2+ is present, but a variety of polynuclear Cu(II) cations have been reported in the literature, including [Cu2(OH)]3+, [Cu2(OH)2]2+, [Cu3(OH)2]4+ and [Cu3(OH)4]2+ (Burgess, 1974), and can be removed by cation exchange columns if necessary.
(c) Control experiment and inferences. Heat alone did not decolourize the solution for the entire length of the heating program, but lightened its colouration.
4.2.5. Decolourisation of the Cr(III)-EBT Dye Solution
The Cr(III)-EBT solution was so intensely wine-red that it was necessary to dilute it 10 times before it could be treated satisfactorily within 1.5 hours by the above means. Therefore, the Cr(III)-EBT dye was the most recalcitrant to this AOP, with peroxodisulphate as the oxidant. When 2 g of Na2S2O8 was added to 100 cm3 of the ten-fold diluted Cr(III)-EBT solution, a reaction solution of pH = 3.2 resulted. Since the solution was 10 times diluted, it was not necessary to furnish all 4 g of the oxidant. The addition of 0.3 g NaOH produced a deep orange solution with pH = 12. The Cr(III)-EBT solution was then subjected to the same heating program as above.
4.2.5.1. Results and Discussion
(a) Observations during decolourisation. The Cr(III)-EBT solution did not decolourise as smoothly as the other metallic complexes encountered so far. The dye solution certainly did not decolourise as soon as the temperature of the bulk of the liquid reached 65°C. However, during its immersion in the water bath at 65°C, the orange colouration gradually turned amber, and then to a transparent solution with a yellowish tinge at the end of the 1-hour period.
The concentration of Cr3+ ions in the 10-times diluted solution was about 8.3 μM (431 ppm). After freeing themselves from EBT dye ligands, Cr3+ ions oxidised to Cr2O72- ions by persulphate in an acidic solution.
After cooling the treated solution to room temperature (at which point pH = 1.70), the liquid was treated further for complete decolourisation.
Step 1: 2 g of activated charcoal was added to the solution and stirred for an hour in a beaker. This adsorption treatment reduced the light yellowish tint further, but it was still detectable by the human eye.
Anionic Cr(VI) species in an aqueous solution can be removed by different types of activated carbon (Selomulya et al., 1999). It is worthwhile to conduct more research on adsorption in a packed bed on a pilot scale with various types of charcoal. The possibility of using a pulsed column (Brunet et al., 2006), whereby mechanical energy is introduced into the packed bed to induce turbulence with the objective of increasing the rate of mass transfer of the dye to the particle surface, should be examined further.
| Metallic ion in untreated dye solution | Final concentration of metal in treated solution (ppm) |
% reduction of metallic content in solution |
Absorbance and appearance of treated solutions |
Extended treatment (the “polishing” step) |
|---|---|---|---|---|
| Ni2+ | 0.053 | 98.9% | 1. Clear, colourless liquid. 2. Absorbance = 0.025 3. Black ppt. likely to be Ni.O(OH) |
Not needed. |
| Co2+ | 0.048 | 99.0% | 1. Clear, colourless liquid. 2. Absorbance = 0.024 3. Black ppt. likely to be Co.O(OH) |
Not needed. |
| Fe2+ | 0.039 | 99.1% | 1. Clear, colourless liquid. 2. Absorbance = 0.022 3. Deep orange ppt. likely to be “Fe(OH)3” |
Not needed. |
| Cu2+ | 0.55 | 89.6% | 1. Light tint of peach colouration. 2. Absorbance = 0.036 after extended treatment. 3. Black CuO ppt. |
Activated charcoal removed the peach tint to give a clear colourless liquid. Ion-exchange removed Cu2+ ions further after acidification. |
| Cr3+ | 1.50 | 65.3% reduction from a 10-fold diluted dye solution | 1. Light yellow tint. 2. Absorbance = 0.047 after extended treatment. 3. No ppt. formed during the entire course of treatment. |
Activated charcoal reduced the vestigial yellow tint further, followed by a reduction of Cr(VI) to Cr(III) with thiosulphate, resulting in colourless water; ion exchange then reduced Cr3+ concentration. |
Step 2: 0.2 g (excess) of the reducing agent sodium thiosulfate Na2S2O3.5H2O was added to reduce the powerful Cr(VI) oxidant in the form of Cr2O72- to the less harmful Cr(III) in the treated solution, which was acidic at this stage. Clear, transparent, and colourless water has been the result. Unpublished work by the authors shows that redox reaction between Cr(VI) and thiosulphate proceeds much faster in an acidic solution than alkaline. It is not advisable to add more than 0.5 g of Na2S2O3.5H2O per 100 cm3 of the solution because the liquid will develop a cloudy white suspension, which will not settle. ICP showed [Cr3+] = 3 ppm at this stage of treatment. The redox reaction between thiosulphate and dichromate is as follows:
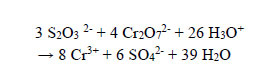 |
(15) |
Cr(III) ions in neutral to slightly acidic media include binuclear species, such as [Cr2O]4+ and [Cr2(OH)2]4+, which are listed by Burgess (1978).
Step 3: Ion-exchange to remove Cr3+ ions was carried out by adding 0.2 g of the “Amberlyst-15” ion-exchange beads and stirring for 30 minutes, resulting in [Cr3+] = 1.5 ppm. The absorbance of the final solution after filtration was 0.047.
No attempt was made to precipitate Cr3+ ions as their hydroxides. Chromium (III) hydroxide re-solubilized due to the oxidation of Cr(III) to soluble Cr(VI) ions in the form of CrO42- in an alkaline solution. This can happen at the surface of the particles, which will initially form the Cr(III) hydroxide colloid (if this pathway is allowed to proceed). The colloidal particles then aggregate to become larger particles (the phenomenon of Smoluchowski coagulation) before sedimentation, but oxidation will not halt.
(b) Chromium recovery. ICP analysis of the final effluent showed residual [Cr3+] = 1.5 ppm (28.9 μM). Recovery of chromium by ion-exchange resins can be calculated by [1 – (28.9/83.3)] x 100% = 65.3%, but this is possible only from the 10-fold diluted solution.
(c) Control experiment and inferences. Heat alone did not diminish the intensity of colouration of the 10-fold diluted solution appreciably. There was no formation of precipitates during heating.
Table 4 presents the reduction of metallic content of all metal-EBT solutions after treatment, the visual observation of these solutions, and the extended treatments required by the Cu(II) and Cr(III) solutions to achieve decolourisation.
Excess persulphate present in all treated wastewaters can be removed in slightly acidic solutions by reaction with thiosulphate; therefore,
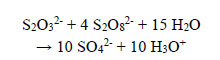 |
(16) |
CONCLUSION
Doubly bonded azo linkages –N=N- are part of the key features, which provide dyes with their unique colours. In this work, aqueous dye solutions of Eriochrome Black T (Mordant Black 11) and of the metal complexes formed by chelation with Ni2+, Co2+, Fe2+, Cu2+ and Cr3+ could be decolourised via redox reactions with sodium peroxodisulphate (Na2S2O8) at 65°C, within 1.5 hours. The concentrations of metallic ions were found to be greatly reduced. The success depended on the concentration of the oxidant, temperature, and duration of the overall reaction, and these are the parameters that should be considered first in the treatment of wastewaters containing metal azo dyes. In none of the control experiments was the action of heat the sole causative factor of decolourisation, although it was observed that heat alone would lighten the intensity of colouration.
The Cr(III)-EBT solution was the most difficult to decolourize, and only after a 10-fold dilution of the prepared dye solution could the processing time be reduced to be similar to that of the other metal-EBT solutions. The techniques of ion exchange, adsorption by activated charcoal, and thiosulphate reduction of a higher oxidation state of a metal to a lower one, such as Cr(VI) to Cr(III), have been shown to be effective in removing the vestiges of colouration; their application can be considered as specific situations demand.
Raman spectroscopy has perhaps demonstrated one important aspect of decolourisation/detoxification to be the fracture of the azo bond -N=N- in the EBT chromophore.
A template for the investigation of wastewater treatment containing metal azo dyes is recommended as a result of this work.
FUTURE WORK
One should decide whether complete (i.e., prolonged) oxidation of fragments of the dye to CO2(g), H2O(l) and NO3-(aq) is necessary for complete detoxification. As part of the EBT molecule, the fate of the sulfonic acid group is unknown and requires investigation by instrumental analysis. The final effluents are to be subjected to the Ames (mutagenicity) test.
From Equations (1), (3), (8) and (9), it can be seen that there are always three powerful oxidants present in aqueous solution, namely, S2O82- (Eo = +2.01 V, SHE); SO4●- (Eo = +2.60 V, SHE); and HO● (Eo = +2.7 V, SHE), although the hydroxyl radical is not expected to be the most abundant compared to the sulfur-centered free radicals encountered in this work. In the literature, it is often stated that the half-life of HO● is extremely short in the vicinity of 1 μs with steady-state concentrations in the low range of 10-12 to 10-10 mol dm-3 in various wastewater treatment plants (Glaze & Kang, 1988; Buffle et al. 2006). It would however be useful to clarify the mechanism to determine or model the relative abundance of all three oxidants at a temperature, such as 65o C (this work), and the rates with which they react with water. This is a fundamental consideration because reactions between oxidants and water are largely sacrificial in any advanced oxidation process (AOP), and these reactions may not contribute to the destruction of the target contaminant in water. However, the question arises as to what percentage of the applied oxidant is sacrificed. This is a significant question concerning operational cost and should be determined by experiment.
LIST OF ABBREVIATION
| EBT | = Eriochrome Black T |
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article are available within the article.
FUNDING
None.
CONFLICT OF INTEREST
Dr. James Barker is on the Editorial Advisory Board of The Open Ecology Journal.
ACKNOWLEDGEMENTS
Declared none.
APPENDIX
“Decolourisation of water-soluble dyes by NaOCl; comparison with Na2S2O8”
Each of the six dye solutions (100 ml each, and each with a dye of concentration 0.1 mM) listed in Table 1 of the main text of this article was mixed with two NaOH pellets (totalling 0.5 gram and bringing the solution pH = 12); the absorbing wavelength and the corresponding absorbance were noted using the UV-visible spectrometer. A volume of 0.5 ml of NaOCl solution (of concentration 2 mol dm-3) was added to each dye solution and absorbance readings were taken every 5 minutes, for an hour. This process was also repeated using 5 ml of NaOCl solution for those dyes that showed no signs of decolourisation after treatment with 0.5 ml NaOCl solution for 1 hour.
Results and Discussion
Only Gentian Violet and Bromophenol Blue were completely decolourised by sodium hypochlorite solution in this set of experiments. The intensity of the colouration of the EBT solution was lessened but was still visible. Methyl Orange, Bromocresol Green and Bromothymol Blue did not seem to respond to treatment and remained intensely coloured. These observations are detailed below.
(1) Gentian Violet
Gentian Violet decolourised immediately on addition of the 0.5 ml NaOCl solution. Due to this very rapid decolourisation, which happened even before the reaction solution could be introduced into the cuvette of the UV-visible spectrometer, no spectra can be taken.
(2) Bromophenol Blue
Bromophenol Blue (also called “BB”) also decolourised completely with 0.5 ml of the NaOCl solution, but unlike Gentian Violet, the decolourisation was not immediate. It occurred near the end of the period of 1 hour of stirring. To rival the speed of decolourisation of Gentian Violet, 20 ml of the NaOCl solution must be used, whereby the intensely blue solution became clear, colourless and transparent within one minute of mixing.
In a separate experiment, a reaction between 0.01 mM (100 ml) Bromophenol Blue (BB) and 0.5 ml of the NaOCl solution was carried out and monitored by tracking and recording the changes in absorbance readings of the visible spectrum at wavelength λ = 591 nm, which did not shift during the reaction. By inspection of the UV-visible spectrum, the absorbances were such that:
At time = 0 min., absorbance (Abs) = 0.340;
At time = 30 min., absorbance (Abs) = 0.16.
The ratio of absorbances 0.16/0.334 = 0.48 ≈ 0.50.
At time = 30 min., absorbance (Abs) = 0.16;
At time = 60 min., absorbance (Abs) = 0.08.
The ratio of absorbances = 0.08/0.16 = 0.50.
It seems that the reaction between Bromophenol Blue (BB) and NaOCl is “pseudo” first order with respect to BB. Therefore, a plot of loge([BB]t=τ / [BB]t=0) vs time, or more specifically (through the application of the Beer-Lambert Law: Absorbance = ε x c x l), a graphical plot of [-loge(Abst=τ / Abst=0)] vs time will reveal whether a straight line relationship exists. If so, the reaction is 1st order with respect to Bromophenol Blue (BB). The graph produced seems to be a straight line, and it can be concluded that the reaction is very likely to be 1st order with respect to the dye Bromophenol Blue, Figure A1.
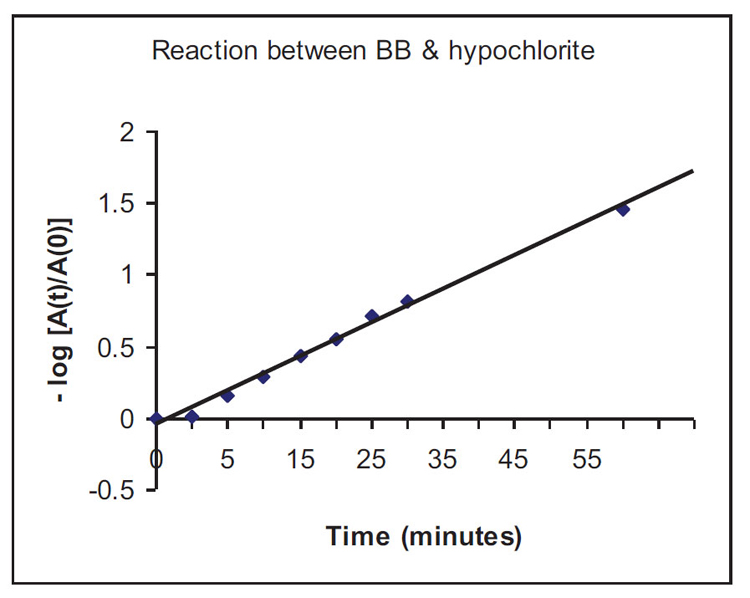
As the half-life of the decolourisation reaction t1/2 ≈ 30 minutes (1800 s), the rate constant of the reaction will be: k = (loge2 / t1/2) = 0.693/1800 = 3.85 x 10-4 s-1. (The same exercise can be repeated for NaOCl by keeping [BB] constant.)
The mole ratio [OCl-]:[dye] = 0.31 / 0.083 x 10-3 = 3735 : 1. These numbers are borne out of experimental trial-and-error with the interesting results presented here. However, it is definitely possible to reduce the use of NaOCl and allow decolourisation to be achieved in a longer time scale (than 1 hour) within the limits of work schedules in industries.
When the same molarity of Marshall’s acid as hypochlorite was used, i.e., 0.31 mol dm-3 (7.4 wt. % solution of the persulphate) at room temp (18°C), no visible changes occurred in 30 minutes. But heating to 60°C resulted in complete decolourisation in 5 minutes. In a separate experiment in which no heat was applied, a concentration of 0.084 mol dm-3 (2 wt. %) Na2S2O8 decolourized EBT at 18°C after 2 weeks. Since the concentration of EBT used was 0.0033 mol dm-3 and that of sodium peroxodisulfate Na2S2O8 used was 0.084 mol dm-3, the minimum working persulfate/EBT ratio is 0.084/0.0033 ≈ 25:1, at room temperature. The slower processes which took place at room temperature allows one to hypothesize that both the one-electron reduction of S2O82- and the homolytic fission of S2O82- are slower than those carried out at elevated temperatures.
(3) Eriochrome Black T (EBT)
In the case of reaction of EBT with NaOCl, the intensity of colouration decreased during the duration of the 1-hour stirring exercise. At t = 0 min., Abs = 1.185; at t = 60 min., Abs = 0.622. Complete decolourisation was not achieved. The interesting observation was the change in the absorbing wavelength half-way through the experiment, between 30 to 35 minutes. At t = 0 min., λ = 531 nm; between t = 30 to 35 min., the absorbing wavelength abruptly switched to λ =395 nm. The solution at this point had a yellow tint that remained unchanged. In a separate experiment, 20 ml of the NaOCl solution was added to a fresh EBT solution and the progress of decolourisation was monitored by visible spectrometry. The deep dark purple colouration disappeared within 1 minute of adding NaOCl, but the same yellow tint remained despite stirring for an hour.
Molarity of EBT dye at the commencement of the experiment = 0.1 x (100/120) mM = 0.083 mM = 0.083 x 10-3 mol dm-3. Molarity of OCl- at beginning of the experiment = 1.86 x (20/120) = 0.31 mol dm-3. The ratio mole ratio [OCl-]:[dye] = 0.31 / 0.083 x 10-3 = 3735:1. When the same molarity of peroxodisulphate as hypochlorite was used, i.e., 0.31 mol dm-3 solution, which means 7.4 wt. % solution, at room temp (18°C), there were no visible changes after 30 minutes of mixing. However, heating the reaction mixture to 60°C completely decolourised the EBT solution in 5 minutes. Again, the relatively slower processes which took place at room temperature allows one to hypothesize that both the one-electron reduction of S2O82- and the homolytic fission of S2O82- are slower than those which took place at higher temperatures.
(4) Methyl Orange, Bromocresol Green and Bromothymol Blue
These dyes remained intensely coloured subsequent to NaOCl treatment. There appeared to be no change in colour at all. In order to discover whether these three dyes will respond to a higher concentration of NaOCl, a separate set of experiments was conducted whereby 5 ml of NaOCl solution was added instead of 0.5 ml, and stirring for 2 hours instead of 1 hour. At the end of the 2 hours, none of the dye solutions had completely decolourised, although the colours were much less intense. Methyl Orange had a light yellow tint not dissimilar to that of the treated EBT solution. The absorbances of the three dyes at the commencement and end of the experiment (at t = 2 hours) are shown in Table A1. It is uncertain whether the yellow tint was a result of excess NaOCl (in which case all the chromophores of the dyes may have been destroyed), or recalcitrant dye fragments with chromophores, or a mixture of both. However, treatment by 7.4 wt% persulphate at 60oC decolourised the dye solutions in 5 minutes. (There were no visible signs of reaction at room temperature). Again, this allows one to hypothesize that redox reactions proceeded faster at higher temperatures.
| Dye | Absorbance (t = 0) | Absorbance (t = 2 hours) |
|---|---|---|
| Methyl Orange | 1.983 | 0.828 |
| Bromocresol Green | 1.755 | 1.406 |
| Bromothymol Blue | 2.586 | 0.552 |
Conclusions
Four types of decolourisation behaviour emerged from these experiments:
1. Gentian Violet and Bromophenol Blue were decolourised with relative ease by NaOCl.
2. The two azo dyes EBT and Methyl Orange left a stubborn yellow tint in solution treated by NaOCl.
3. Solutions of Bromocresol Green and Bromothymol Blue remained intensely coloured subsequent to treatment by NaOCl.
4. The only set of experimental conditions which rendered satisfactory decolourisation of dyes is the use of peroxodisulphate as an oxidant at the concentration of 7.4 wt% and 60oC; decolourisations were achieved within 5 minutes of reaction. This also allows one to hypothesize that the one-electron reduction of S2O82- and homolytic thermolysis of S2O82-, or both, can only be deployed at an elevated temperature such as 60oC. Both of these reactions may proceed at a much slower rate at room temperatures.
REFERENCES
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link] [PubMed Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link] [PubMed Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link] [PubMed Link]
[CrossRef Link] [PubMed Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link]
[CrossRef Link] [PubMed Link]

